Increased Nucleophilicity of Heavier Nucleophiles (Section 1.6.1.1, p. 28).
I stated that heavier atoms are more nucleophilic because they are more polarizable. This argument is very common, but there is evidence that it is not valid. I- is indeed much more nucleophilic than Cl- in MeOH (the traditional solvent for measuring nucleophilicity), but in aprotic solvents and in the gas phase, Cl- is slightly more nucleophilic than I-. If nucleophilicity were determined by polarizability, then it would not be solvent-dependent.
In the case of anions (e.g., I- vs. Cl-), the bext explanation for the nucleophilicity trend is that protic solvents such as MeOH can hydrogen-bond more strongly to smaller anions, rendering them less nucleophilic. In solvents where hydrogen-bonding is not possible, smaller anions are both more nucleophilic and more basic than larger ones. In the case of neutral nucleophiles (e.g., Et2S vs. Et2O), both the smaller electronegativity of heavier atoms and their greater steric accessibility (because of the longer bond lengths) explain their greater nucleophilicity.
The second line of the example is incorrect. The C-Cl σ bond in the first two structures in the second line of the example should be replaced with a C=O π bond. The correct mechanism is:
Thanks to Markus "Eagle-Eyes" Knobloch for spotting this error.
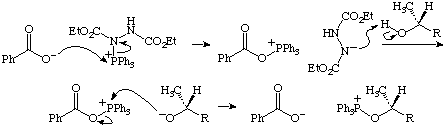
In the second step of the given mechanism, the carboxylate deprotonates the alcohol, which then attacks the phosphoniohydrazine to displace N from P. The book notes that the deprotonation step violates the pKa rule (pKa alcohol = 17, pKb of carboxylate = 5). A better alternative has the carboxylate displace N from P in SN2 fashion, giving an acyloxyphosphonium ion and an nitrogen anion. The latter then deprotonates the alcohol, generating an alkoxide, which displaces P from O to give the requisite alkoxyphosphonium ion and to regenerate the carboxylate.
Diazonium ions are said to react with nucleophiles by an SN1-type mechanism (via a phenyl cation), but better mechanisms can be drawn for most nucleophiles. For example, I- and H3PO2 are thought to react by an SRN1 mechanism, whereas CuX nucleophiles are thought to react by a nonchain electron transfer mechanism. Either of these mechanisms allows one to avoid drawing a ridiculously high energy phenyl cation, which should be avoided if at all possible. The SN1-type mechanism is reasonable only when H2O or BF4- are the nucleophiles, producing a phenol or an aryl fluoride. Thanks to Prof. Carlo Galli for bringing these points to my attention.
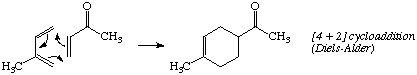
The product of the [4 + 2] cycloaddition at the top of the page is incorrect. The correct product is the 1,4-disubstituted compound.
Thanks to Sihui Long for spotting this error.
Danishefsky's diene is 1-methoxy-3-trimethylsilyloxy-1,3-butadiene, not 1-trimethylsilyloxy-3-methoxy-1,3-butadiene. The positions of the Me3SiO and MeO groups in the second structure in the first graphic on p. 164 should be switched. Thanks to Markus "Eagle-Eyes" Knobloch for spotting this error.
The third structure in the mechanism towards the bottom of p. 177 contains a pentavalent N atom. The +N=O should be +N-O-. Thanks to James Kirkham for spotting this error.
The Wittig reaction is said to be allowed by the Woodward-Hoffmann rules because of the participation of a d orbital on the P atom, which makes the Wittig reagent's HOMO antisymmetric. This explanation has appeared in the literature for many, many years, but calculations have shown that the d orbital of a P atom is much too high in energy to participate in a π bond with a C(p) orbital. Instead, the π bond is made up of a C(p) orbital and a C-P σ* orbital (hyperconjugation).
Why, then, does the Wittig reaction proceed, even though it violates the Woodward-Hoffmann rules? The Woodward-Hoffmann rules say that the symmetries of the HOMO and LUMO of the reagents in a pericyclic reaction have a strong effect on the TS energy. In the TS of a pericyclic reaction involving compounds with "normal" π bonds, there are strong interactions among orbitals both between the reactants and within each reactant. In P=C π bonds, however, the overlap between the constituent orbitals is much poorer, and so the influence of symmetry on the reaction is much weaker. In this case the strength of the bonds being made and broken is much more important than symmetry, and the reaction proceeds. The same is true of the retrocycloaddition.
Another way of thinking of this problem: Every π bond can also be written as a 1,2-diradical. This resonance structure is very important for P=C π bonds. Symmetry influences TS energy only through the π bond resonance structure. Because there is such a strong 1,2-diradical component in P=C π bonds, there is only a small increase in energy due to mismatched symmetry upon cycloaddition of the P=C π bond and a C=O π bond, and it is far outweighed by the energy gained by forming new P-O and C-C σ bonds. The same is true of the retrocycloaddition.
Thanks to Prof. Roger Grev for explaining these concepts to me.
The preference for endo adducts in Diels-Alder reactions and other cycloadditions is said to be due to secondary orbital interactions in the TS for the cycloaddition. Recent investigations have shown that the endo selectivity in many reactions can be explained by hydrophobic, steric, and electrostatic effects. Secondary orbital interactions do not have to be invoked.
Presumably similar explanations can be invoked for the endo selectivities observed for [3 + 2] and [2 + 2] cycloadditions. See José I. García, José A. Mayoral, and Luis Salvatella, Acc. Chem. Res. 2000 33 (10), 658 for a fuller discussion of these issues. Please note, though, that these ideas are new, and a scientific consensus has not been established.
Thanks to Prof. Luis Salvatella for pointing out these observations.
A line of graphics is missing from the mechanism, which is correctly drawn as follows:
There is an error in the second step of the propagation. It should read as follows:
Thanks to Aaron Skaggs for alerting me to this error.
Fe(0) is d8, not d10, and it has six 3d electrons, not eight. Thanks to Freddie Hughes, Jr. for alerting me to this error.
The number of CO ligands is given incorrectly. The problem should read as follows:
Thanks to Markus "Eagle-Eyes" Knobloch for spotting this error.
The last sentence of the second paragraph should read,
Thanks to Francisco Velazquez for alerting me to this error.
The third sentence of the fourth paragraph should read,
Thanks to Francisco Velazquez for alerting me to this error.
Return to the home page for AWRORM.
This page was last updated
Example of the Benzylic Acid Rearrangement (Section 2.5.1, p. 83).
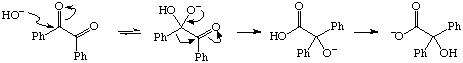
Mechanism of the Mitsunobu Reaction (Section 2.6.2, p. 89).
Aromatic Substitution of Anilines via Diazonium Salts (Section 3.4.2, pp. 121-122).
Examples of Cycloadditions (Section 4.1.1, p. 141).
Good Diels-Alder Dienes (Section 4.3.1.1, pp. 163-164).
Pentavalent N in nitrile oxide mechanism (p. 168)
Stereospecificity of the Wittig reaction (Section 4.3.3, p. 179).
Origin of the Endo Rule (Section 4.3.4, p. 182).
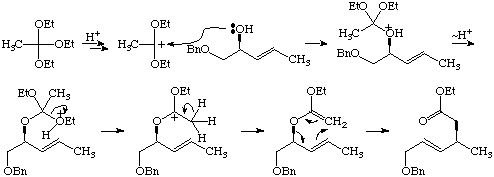
Mechanism of Orthoester Claisen Rearrangement (Section 4.4.1, p. 188).
Mechanism of Dehalogenation (Section 5.2, p. 228).
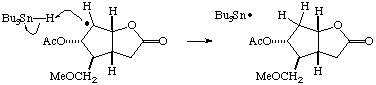
Oxidation state and d Electron Count (Section 6.1.1.2, p. 261).
Problem 6.1 (Section 6.2.1.6, p. 275).
Problem 6.1. NMO oxidizes one CO ligand of the alkyne-Co2(CO)6 complex to CO2 and gives an alkyne-Co2(CO)5 complex. Write a mechanism for this transformation.
Introduction to Metal-Catalyzed Reactions (Section 6.3, p. 279).
Often the number and nature of the ligands on the metal are unknown, and so the metal center and its associated ligands are indicated merely as LnM.
Late-Metal-Catalyzed Hydrogenations and Hydrometallation (Section 6.3.1.1, p. 280).
β-Hydride elimination then gives H—Pd(II)—H and CO2.